以下文章转载自健康报。
这是一种奇怪的病症,在很长一段时间内,医学界都对它攻而不克,只能维持治疗,无法彻底治愈。更有医生断言:这个病如果重度的话,多在5岁前就会死亡,一般不会活过20岁。这就是地中海贫血症(简称地贫),因为早期发病人群多为地中海沿岸居民而得名。这是一种遗传性很强的疾病,父母携带致病基因,很有可能殃及孩子。
2015年,中国地贫联盟等机构联合发布的《中国地中海贫血蓝皮书》显示,我国长江以南地区地贫较为集中,尤以两广地区最为严重。很多重度地贫患者从出生开始,就进入了生命倒计时,而医学界的不断努力,正是与死神在较量。
治疗手段有限医疗花费巨大一段20多年前的地中海贫血症宣传片中,年仅14岁的少年面对镜头介绍自己的生活状态:“别人放学都去打球,我一放学就要去输血,不输血是会死的。”面带笑容的他说出这段话时,让人感到心酸。
这就是地中海贫血患者的真实生存状态:永远徘徊在生与死的边缘。绝大部分患者从确诊的那一天起,生命的主动权就不在自己手里了。
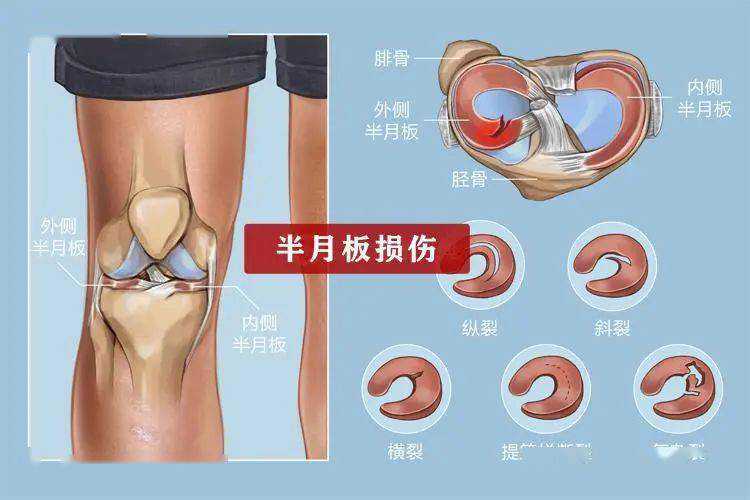
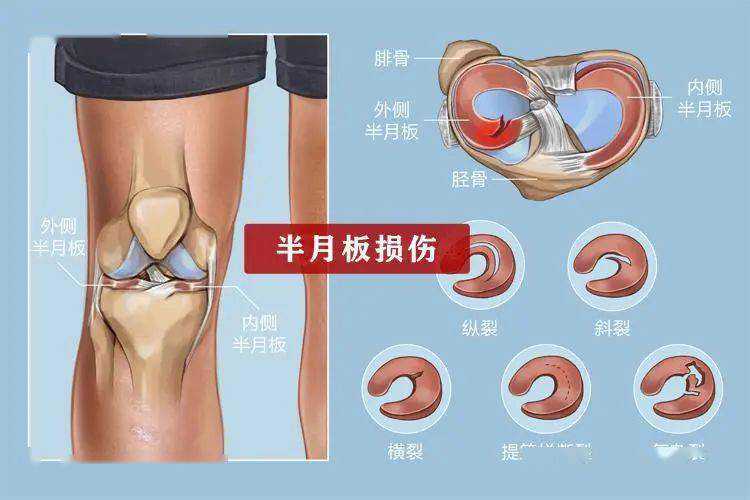
地中海贫血症按照氨基酸链来分类,通常可以分为α、β、δβ和δ等4种类型,其中以β和α地中海贫血症较为常见。β地中海贫血症是由位于第11号染色体的β-珠蛋白基因决定簇发生遗传性点突变或缺失,β-珠蛋白合成完全或部分受抑制,导致α/β链比例失衡,从而产生一系列症状和体征的遗传性溶血性疾病。
重度β地贫,又称Cooley贫血,患儿出生时无症状,3到6个月开始发病,呈现慢性进行性贫血症状,会表现出面色苍白、肝脾肿大、发育不良、轻度黄疸等一系列症状,并具有典型的地中海贫血特殊面容。随着年龄的增长,患者会并发含铁血黄素沉着症,引发多个脏器损害,最终患者会因为心力衰竭而死亡。
在中国,目前只有除铁药物和规律输血才能缓解地贫的症状,但不是每个人都有条件接受这样的治疗方案,因为常规除铁药物每年的花费约为10万元,终生输血更是一笔庞大的开支。大多数家庭无法承受这样的沉重负担。
造血干细胞移植(HSCT)是目前证明能治疗重度地贫患儿的唯一方法,但俗称“骨髓移植”的这一疗法除了花费巨大外,找到合适的配型也不容易。
基因疗法不需异体移植地贫已经成为人口出生缺陷的世界公共卫生问题。面对疾病的挑战,医生和科学家们一直在探索治疗地贫的有效方法。
2019年11月8日,美国新基制药的β地中海贫血药物Reblozyl,因为获得了FDA认定的孤儿药资格,得以快速获批上市。
在此之前的6月14日,欧盟也批准了美国蓝鸟生物(bluebirdbio)基因疗法Zynteglo,用于治疗12岁及以上的非β0/β0基因型输血依赖性β地中海贫血患者。蓝鸟生物最新公布的数据显示,患者可以超过1年不进行输血治疗。
这两种治疗方案是完全不同的两条治疗路径。Reblozyl是一款创新型红细胞成熟剂(EMA),可促进动物模型中晚期红细胞的成熟,减少患者所需输血量;Zynteglo则是通过基因编辑技术,从患者体内分离出造血干细胞,并利用病毒载体引入经过修饰、拥有正常功能的基因,利用患者自身的干细胞实现治疗的目的,不需要异体移植。
事实上,国内也有类似的基因疗法正在研发。博雅辑因(北京)生物科技有限公司采取了和蓝鸟生物不同的CRISPR/Cas9基因编辑技术,在精准位点上进行基因修复,而不是引入基因,避免了插入突变的安全性问题。目前,博雅辑因的地中海贫血基因编辑疗法产品已实现了临床级规模化生产,并将于近期公布其体外及体内临床前安全性、有效性数据。
说到基因编辑,人们难免会联想到2018年底,震惊学术界的“基因编辑婴儿”事件。由于“基因编辑婴儿”涉及对生殖细胞和胚胎的改造,遭遇“改造人”的伦理问题。为此,2019年3月14日,《自然》杂志刊发了来自7个国家的科学家和伦理学家以及美国国立卫生研究院的呼吁,要求暂停所有用于临床的人类生殖细胞编辑,并建立国际监管框架。
或许正如一些地贫患者在病友群里说的:“基因疗法是中国数万名重症地贫患者的希望。”作为一种罕见病的解决方案,基因疗法尽早进入实质性临床,将是患者之幸。

健康报